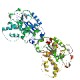
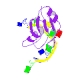
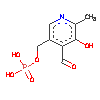
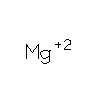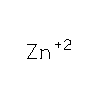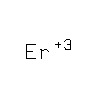How an exonuclease decides where to
stop in trimming of nucleic acids: crystal structures of RNase
T-product complexes
(2012) Nucleic Acids Res. [ Display Full Abstract
| Display for All Results
]
How an exonuclease decides where to
stop in trimming of nucleic acids: crystal structures of RNase
T-product complexes
(2012) Nucleic Acids Res. [ Display Full Abstract
| Display for All Results
]
How an exonuclease decides where to
stop in trimming of nucleic acids: crystal structures of RNase
T-product complexes
(2012) Nucleic Acids Res. [ Display Full Abstract
| Display for All Results
]
How an exonuclease decides where to
stop in trimming of nucleic acids: crystal structures of RNase
T-product complexes
(2012) Nucleic Acids Res. [ Display Full Abstract
| Display for All Results
]
How an exonuclease decides where to
stop in trimming of nucleic acids: crystal structures of RNase
T-product complexes
(2012) Nucleic Acids Res. [ Display Full Abstract
| Display for All Results
]
How an exonuclease decides where to
stop in trimming of nucleic acids: crystal structures of RNase
T-product complexes
(2012) Nucleic Acids Res. [ Display Full Abstract
| Display for All Results
]
How an exonuclease decides where to
stop in trimming of nucleic acids: crystal structures of RNase
T-product complexes
(2012) Nucleic Acids Res. [ Display Full Abstract
| Display for All Results
]
Structural basis for RNA trimming by
RNase T in stable RNA 3'-end maturation
(2011) Nat.Chem.Biol.7:
236-243
PubMed Abstract:
RNA maturation relies on various exonucleases to remove nucleotides
successively from the 5' or 3' end of nucleic acids. However, little is
known regarding the molecular basis for substrate and cleavage
preference of exonucleases. Our biochemical and structural analyses on
RNase T-DNA complexes show that the RNase T dimer has an ideal
architecture for binding a duplex with a short 3' overhang to produce a
digestion product of a duplex with a 2-nucleotide (nt) or 1-nt 3'
overhang, depending on the composition of the last base pair in the
duplex. A 'C-filter' in RNase T screens out the nucleic acids with
3'-terminal cytosines for hydrolysis by inducing a disruptive
conformational change at the active site. Our results reveal the
general principles and the working mechanism for the final trimming
step made by RNase T in the maturation of stable RNA and pave the way
for the understanding of other DEDD family exonucleases.
Structural basis for RNA trimming by
RNase T in stable RNA 3'-end maturation
(2011) Nat.Chem.Biol.7:
236-243
PubMed Abstract:
RNA maturation relies on various exonucleases to remove nucleotides
successively from the 5' or 3' end of nucleic acids. However, little is
known regarding the molecular basis for substrate and cleavage
preference of exonucleases. Our biochemical and structural analyses on
RNase T-DNA complexes show that the RNase T dimer has an ideal
architecture for binding a duplex with a short 3' overhang to produce a
digestion product of a duplex with a 2-nucleotide (nt) or 1-nt 3'
overhang, depending on the composition of the last base pair in the
duplex. A 'C-filter' in RNase T screens out the nucleic acids with
3'-terminal cytosines for hydrolysis by inducing a disruptive
conformational change at the active site. Our results reveal the
general principles and the working mechanism for the final trimming
step made by RNase T in the maturation of stable RNA and pave the way
for the understanding of other DEDD family exonucleases.
Structural basis for RNA trimming by
RNase T in stable RNA 3'-end maturation
(2011) Nat.Chem.Biol.7:
236-243
PubMed Abstract:
RNA maturation relies on various exonucleases to remove nucleotides
successively from the 5' or 3' end of nucleic acids. However, little is
known regarding the molecular basis for substrate and cleavage
preference of exonucleases. Our biochemical and structural analyses on
RNase T-DNA complexes show that the RNase T dimer has an ideal
architecture for binding a duplex with a short 3' overhang to produce a
digestion product of a duplex with a 2-nucleotide (nt) or 1-nt 3'
overhang, depending on the composition of the last base pair in the
duplex. A 'C-filter' in RNase T screens out the nucleic acids with
3'-terminal cytosines for hydrolysis by inducing a disruptive
conformational change at the active site. Our results reveal the
general principles and the working mechanism for the final trimming
step made by RNase T in the maturation of stable RNA and pave the way
for the understanding of other DEDD family exonucleases.
Structural basis for RNA trimming by
RNase T in stable RNA 3'-end maturation
(2011) Nat.Chem.Biol.7:
236-243
PubMed Abstract:
RNA maturation relies on various exonucleases to remove nucleotides
successively from the 5' or 3' end of nucleic acids. However, little is
known regarding the molecular basis for substrate and cleavage
preference of exonucleases. Our biochemical and structural analyses on
RNase T-DNA complexes show that the RNase T dimer has an ideal
architecture for binding a duplex with a short 3' overhang to produce a
digestion product of a duplex with a 2-nucleotide (nt) or 1-nt 3'
overhang, depending on the composition of the last base pair in the
duplex. A 'C-filter' in RNase T screens out the nucleic acids with
3'-terminal cytosines for hydrolysis by inducing a disruptive
conformational change at the active site. Our results reveal the
general principles and the working mechanism for the final trimming
step made by RNase T in the maturation of stable RNA and pave the way
for the understanding of other DEDD family exonucleases.
Structural basis for RNA trimming by
RNase T in stable RNA 3'-end maturation
(2011) Nat.Chem.Biol.7:
236-243
PubMed Abstract:
RNA maturation relies on various exonucleases to remove nucleotides
successively from the 5' or 3' end of nucleic acids. However, little is
known regarding the molecular basis for substrate and cleavage
preference of exonucleases. Our biochemical and structural analyses on
RNase T-DNA complexes show that the RNase T dimer has an ideal
architecture for binding a duplex with a short 3' overhang to produce a
digestion product of a duplex with a 2-nucleotide (nt) or 1-nt 3'
overhang, depending on the composition of the last base pair in the
duplex. A 'C-filter' in RNase T screens out the nucleic acids with
3'-terminal cytosines for hydrolysis by inducing a disruptive
conformational change at the active site. Our results reveal the
general principles and the working mechanism for the final trimming
step made by RNase T in the maturation of stable RNA and pave the way
for the understanding of other DEDD family exonucleases.
Structural and biochemical
characterization of CRN-5 and Rrp46: an exosome component participating
in apoptotic DNA degradation
(2010) Rna16: 1748-1759
PubMed Abstract:
Rrp46 was first identified as a protein component of the eukaryotic
exosome, a protein complex involved in 3' processing of RNA during RNA
turnover and surveillance. The Rrp46 homolog, CRN-5, was subsequently
characterized as a cell death-related nuclease, participating in DNA
fragmentation during apoptosis in Caenorhabditis elegans. Here we
report the crystal structures of CRN-5 and rice Rrp46 (oRrp46) at a
resolution of 3.9 A and 2.0 A, respectively. We found that recombinant
human Rrp46 (hRrp46), oRrp46, and CRN-5 are homodimers, and that
endogenous hRrp46 and oRrp46 also form homodimers in a cellular
environment, in addition to their association with a protein complex.
Dimeric oRrp46 had both phosphorolytic RNase and hydrolytic DNase
activities, whereas hRrp46 and CRN-5 bound to DNA without detectable
nuclease activity. Site-directed mutagenesis in oRrp46 abolished either
its DNase (E160Q) or RNase (K75E/Q76E) activities, confirming the
critical importance of these residues in catalysis or substrate
binding. Moreover, CRN-5 directly interacted with the apoptotic
nuclease CRN-4 and enhanced the DNase activity of CRN-4, suggesting
that CRN-5 cooperates with CRN-4 in apoptotic DNA degradation. Taken
together all these results strongly suggest that Rrp46 forms a
homodimer separately from exosome complexes and, depending on species,
is either a structural or catalytic component of the machinery that
cleaves DNA during apoptosis.
Structural and biochemical
characterization of CRN-5 and Rrp46: an exosome component participating
in apoptotic DNA degradation
(2010) Rna16: 1748-1759
PubMed Abstract:
Rrp46 was first identified as a protein component of the eukaryotic
exosome, a protein complex involved in 3' processing of RNA during RNA
turnover and surveillance. The Rrp46 homolog, CRN-5, was subsequently
characterized as a cell death-related nuclease, participating in DNA
fragmentation during apoptosis in Caenorhabditis elegans. Here we
report the crystal structures of CRN-5 and rice Rrp46 (oRrp46) at a
resolution of 3.9 A and 2.0 A, respectively. We found that recombinant
human Rrp46 (hRrp46), oRrp46, and CRN-5 are homodimers, and that
endogenous hRrp46 and oRrp46 also form homodimers in a cellular
environment, in addition to their association with a protein complex.
Dimeric oRrp46 had both phosphorolytic RNase and hydrolytic DNase
activities, whereas hRrp46 and CRN-5 bound to DNA without detectable
nuclease activity. Site-directed mutagenesis in oRrp46 abolished either
its DNase (E160Q) or RNase (K75E/Q76E) activities, confirming the
critical importance of these residues in catalysis or substrate
binding. Moreover, CRN-5 directly interacted with the apoptotic
nuclease CRN-4 and enhanced the DNase activity of CRN-4, suggesting
that CRN-5 cooperates with CRN-4 in apoptotic DNA degradation. Taken
together all these results strongly suggest that Rrp46 forms a
homodimer separately from exosome complexes and, depending on species,
is either a structural or catalytic component of the machinery that
cleaves DNA during apoptosis.
Crystal structure of CRN-4:
implications for domain function in apoptotic DNA degradation
(2009) Mol.Cell.Biol.29:
448-457
PubMed Abstract:
Cell death related nuclease 4 (CRN-4) is one of the apoptotic nucleases
involved in DNA degradation in Caenorhabditis elegans. To understand
how CRN-4 is involved in apoptotic DNA fragmentation, we analyzed
CRN-4's biochemical properties, in vivo cell functions, and the crystal
structures of CRN-4 in apo-form, Mn(2+)-bound active form, and
Er(3+)-bound inactive form. CRN-4 is a dimeric nuclease with the
optimal enzyme activity in cleaving double-stranded DNA in apoptotic
salt conditions. Both mutational studies and the structures of the
Mn(2+)-bound CRN-4 revealed the geometry of the functional nuclease
active site in the N-terminal DEDDh domain. The C-terminal domain,
termed the Zn-domain, contains basic surface residues ideal for nucleic
acid recognition and is involved in DNA binding, as confirmed by
deletion assays. Cell death analysis in C. elegans further demonstrated
that both the nuclease active site and the Zn-domain are required for
crn-4's function in apoptosis. Combining all of the data, we suggest a
structural model where chromosomal DNA is bound at the Zn-domain and
cleaved at the DEDDh nuclease domain in CRN-4 when the cell is
undergoing apoptosis.
Crystal structure of CRN-4:
implications for domain function in apoptotic DNA degradation
(2009) Mol.Cell.Biol.29:
448-457
PubMed Abstract:
Cell death related nuclease 4 (CRN-4) is one of the apoptotic nucleases
involved in DNA degradation in Caenorhabditis elegans. To understand
how CRN-4 is involved in apoptotic DNA fragmentation, we analyzed
CRN-4's biochemical properties, in vivo cell functions, and the crystal
structures of CRN-4 in apo-form, Mn(2+)-bound active form, and
Er(3+)-bound inactive form. CRN-4 is a dimeric nuclease with the
optimal enzyme activity in cleaving double-stranded DNA in apoptotic
salt conditions. Both mutational studies and the structures of the
Mn(2+)-bound CRN-4 revealed the geometry of the functional nuclease
active site in the N-terminal DEDDh domain. The C-terminal domain,
termed the Zn-domain, contains basic surface residues ideal for nucleic
acid recognition and is involved in DNA binding, as confirmed by
deletion assays. Cell death analysis in C. elegans further demonstrated
that both the nuclease active site and the Zn-domain are required for
crn-4's function in apoptosis. Combining all of the data, we suggest a
structural model where chromosomal DNA is bound at the Zn-domain and
cleaved at the DEDDh nuclease domain in CRN-4 when the cell is
undergoing apoptosis.
Crystal structure of CRN-4:
implications for domain function in apoptotic DNA degradation
(2009) Mol.Cell.Biol.29:
448-457
PubMed Abstract:
Cell death related nuclease 4 (CRN-4) is one of the apoptotic nucleases
involved in DNA degradation in Caenorhabditis elegans. To understand
how CRN-4 is involved in apoptotic DNA fragmentation, we analyzed
CRN-4's biochemical properties, in vivo cell functions, and the crystal
structures of CRN-4 in apo-form, Mn(2+)-bound active form, and
Er(3+)-bound inactive form. CRN-4 is a dimeric nuclease with the
optimal enzyme activity in cleaving double-stranded DNA in apoptotic
salt conditions. Both mutational studies and the structures of the
Mn(2+)-bound CRN-4 revealed the geometry of the functional nuclease
active site in the N-terminal DEDDh domain. The C-terminal domain,
termed the Zn-domain, contains basic surface residues ideal for nucleic
acid recognition and is involved in DNA binding, as confirmed by
deletion assays. Cell death analysis in C. elegans further demonstrated
that both the nuclease active site and the Zn-domain are required for
crn-4's function in apoptosis. Combining all of the data, we suggest a
structural model where chromosomal DNA is bound at the Zn-domain and
cleaved at the DEDDh nuclease domain in CRN-4 when the cell is
undergoing apoptosis.
Structural insights into TDP-43 in
nucleic-acid binding and domain interactions
(2009) Nucleic Acids Res.37:
1799-1808
PubMed Abstract:
TDP-43 is a pathogenic protein: its normal function in binding to
UG-rich RNA is related to cystic fibrosis, and inclusion of its
C-terminal fragments in brain cells is directly linked to
frontotemporal lobar degeneration (FTLD) and amyotrophic lateral
sclerosis (ALS). Here we report the 1.65 A crystal structure of the
C-terminal RRM2 domain of TDP-43 in complex with a single-stranded DNA.
We show that TDP-43 is a dimeric protein with two RRM domains, both
involved in DNA and RNA binding. The crystal structure reveals the
basis of TDP-43's TG/UG preference in nucleic acids binding. It also
reveals that RRM2 domain has an atypical RRM-fold with an additional
beta-strand involved in making protein-protein interactions. This self
association of RRM2 domains produced thermal-stable RRM2 assemblies
with a melting point greater than 85 degrees C as monitored by circular
dichroism at physiological conditions. These studies thus characterize
the recognition between TDP-43 and nucleic acids and the mode of RRM2
self association, and provide molecular models for understanding the
role of TDP-43 in cystic fibrosis and the neurodegenerative diseases
related to TDP-43 proteinopathy.
Crystal structure of human
polynucleotide phosphorylase: insights into its domain function in RNA
binding and degradation
(2011) Nucleic Acids Res. [ Display Full Abstract
| Display for All Results
]
Crystal structure of Escherichia
coli PNPase: central channel residues are involved in processive RNA
degradation.
(2008) Rna14: 2361-2371
PubMed Abstract:
Bacterial polynucleotide phosphorylase (PNPase) plays a major role in
mRNA turnover by the degradation of RNA from the 3'- to 5'-ends. Here,
we determined the crystal structures of the wild-type and a C-terminal
KH/S1 domain-truncated mutant (DeltaKH/S1) of Escherichia coli PNPase
at resolutions of 2.6 A and 2.8 A, respectively. The six RNase PH
domains of the trimeric PNPase assemble into a ring-like structure
containing a central channel. The truncated mutant DeltaKH/S1 bound and
cleaved RNA less efficiently with an eightfold reduced binding
affinity. Thermal melting and acid-induced trimer dissociation studies,
analyzed by circular dichroism and dynamic light scattering, further
showed that DeltaKH/S1 formed a less stable trimer than the full-length
PNPase. The crystal structure of DeltaKH/S1 is more expanded,
containing a slightly wider central channel than that of the wild-type
PNPase, suggesting that the KH/S1 domain helps PNPase to assemble into
a more compact trimer, and it regulates the channel size
allosterically. Moreover, site-directed mutagenesis of several arginine
residues in the channel neck regions produced defective PNPases that
either bound and cleaved RNA less efficiently or generated longer
cleaved oligonucleotide products, indicating that these arginines were
involved in RNA binding and processive degradation. Taking these
results together, we conclude that the constricted central channel and
the basic-charged residues in the channel necks of PNPase play crucial
roles in trapping RNA for processive exonucleolytic degradation.
Crystal structure of Escherichia
coli PNPase: central channel residues are involved in processive RNA
degradation.
(2008) Rna14: 2361-2371
PubMed Abstract:
Bacterial polynucleotide phosphorylase (PNPase) plays a major role in
mRNA turnover by the degradation of RNA from the 3'- to 5'-ends. Here,
we determined the crystal structures of the wild-type and a C-terminal
KH/S1 domain-truncated mutant (DeltaKH/S1) of Escherichia coli PNPase
at resolutions of 2.6 A and 2.8 A, respectively. The six RNase PH
domains of the trimeric PNPase assemble into a ring-like structure
containing a central channel. The truncated mutant DeltaKH/S1 bound and
cleaved RNA less efficiently with an eightfold reduced binding
affinity. Thermal melting and acid-induced trimer dissociation studies,
analyzed by circular dichroism and dynamic light scattering, further
showed that DeltaKH/S1 formed a less stable trimer than the full-length
PNPase. The crystal structure of DeltaKH/S1 is more expanded,
containing a slightly wider central channel than that of the wild-type
PNPase, suggesting that the KH/S1 domain helps PNPase to assemble into
a more compact trimer, and it regulates the channel size
allosterically. Moreover, site-directed mutagenesis of several arginine
residues in the channel neck regions produced defective PNPases that
either bound and cleaved RNA less efficiently or generated longer
cleaved oligonucleotide products, indicating that these arginines were
involved in RNA binding and processive degradation. Taking these
results together, we conclude that the constricted central channel and
the basic-charged residues in the channel necks of PNPase play crucial
roles in trapping RNA for processive exonucleolytic degradation.
Structural and functional insights
into human Tudor-SN, a key component linking RNA interference and
editing.
(2008) Nucleic Acids Res.36:
3579-3589
PubMed Abstract:
Human Tudor-SN is involved in the degradation of hyper-edited
inosine-containing microRNA precursors, thus linking the pathways of
RNA interference and editing. Tudor-SN contains four tandem repeats of
staphylococcal nuclease-like domains (SN1-SN4) followed by a tudor and
C-terminal SN domain (SN5). Here, we showed that Tudor-SN requires
tandem repeats of SN domains for its RNA binding and cleavage activity.
The crystal structure of a 64-kD truncated form of human Tudor-SN
further shows that the four domains, SN3, SN4, tudor and SN5, assemble
into a crescent-shaped structure. A concave basic surface formed
jointly by SN3 and SN4 domains is likely involved in RNA binding, where
citrate ions are bound at the putative RNase active sites. Additional
modeling studies provide a structural basis for Tudor-SN's preference
in cleaving RNA containing multiple I.U wobble-paired sequences.
Collectively, these results suggest that tandem repeats of SN domains
in Tudor-SN function as a clamp to capture RNA substrates.
The Conserved Asparagine in the Hnh
Motif Serves an Important Structural Role in Metal Finger Endonucleases.
(2007) J.Mol.Biol.368: 812
PubMed Abstract:
The HNH motif is a small nucleic acid binding and cleavage module,
widespread in metal finger endonucleases in all life kingdoms. Here we
studied a non-specific endonuclease, the nuclease domain of ColE7
(N-ColE7), to decipher the role of the conserved asparagine and
histidine residues in the HNH motif. We found, using fluorescence
resonance energy transfer (FRET) assays, that the DNA hydrolysis
activity of H545 N-ColE7 mutants was completely abolished while
activities of N560 and H573 mutants varied from 6.9% to 83.2% of the
wild-type activity. The crystal structures of three N-ColE7 mutants in
complex with the inhibitor Im7, N560A-Im7, N560D-Im7 and H573A-Im7,
were determined at a resolution of 1.9 A to 2.2 A. H573 is responsible
for metal ion binding in the wild-type protein, as the zinc ion is
still partially associated in the structure of H573A, suggesting that
H573 plays a supportive role in metal binding. Both N560A and N560D
contain a disordered loop in the HNH motif due to the disruption of the
hydrogen bond network surrounding the side-chain of residue 560, and as
a result, the imidazole ring of the general base residue H545 is tilted
slightly and the scissile phosphate is shifted, leading to the large
reductions in hydrolysis activities. These results suggest that the
highly conserved asparagine in the HNH motif, in general, plays a
structural role in constraining the loop in the metal finger structure
and keeping the general base histidine and scissile phosphate in the
correct position for DNA hydrolysis.
The Conserved Asparagine in the Hnh
Motif Serves an Important Structural Role in Metal Finger Endonucleases.
(2007) J.Mol.Biol.368: 812
PubMed Abstract:
The HNH motif is a small nucleic acid binding and cleavage module,
widespread in metal finger endonucleases in all life kingdoms. Here we
studied a non-specific endonuclease, the nuclease domain of ColE7
(N-ColE7), to decipher the role of the conserved asparagine and
histidine residues in the HNH motif. We found, using fluorescence
resonance energy transfer (FRET) assays, that the DNA hydrolysis
activity of H545 N-ColE7 mutants was completely abolished while
activities of N560 and H573 mutants varied from 6.9% to 83.2% of the
wild-type activity. The crystal structures of three N-ColE7 mutants in
complex with the inhibitor Im7, N560A-Im7, N560D-Im7 and H573A-Im7,
were determined at a resolution of 1.9 A to 2.2 A. H573 is responsible
for metal ion binding in the wild-type protein, as the zinc ion is
still partially associated in the structure of H573A, suggesting that
H573 plays a supportive role in metal binding. Both N560A and N560D
contain a disordered loop in the HNH motif due to the disruption of the
hydrogen bond network surrounding the side-chain of residue 560, and as
a result, the imidazole ring of the general base residue H545 is tilted
slightly and the scissile phosphate is shifted, leading to the large
reductions in hydrolysis activities. These results suggest that the
highly conserved asparagine in the HNH motif, in general, plays a
structural role in constraining the loop in the metal finger structure
and keeping the general base histidine and scissile phosphate in the
correct position for DNA hydrolysis.
The Conserved Asparagine in the Hnh
Motif Serves an Important Structural Role in Metal Finger Endonucleases.
(2007) J.Mol.Biol.368: 812-821
PubMed Abstract:
The HNH motif is a small nucleic acid binding and cleavage module,
widespread in metal finger endonucleases in all life kingdoms. Here we
studied a non-specific endonuclease, the nuclease domain of ColE7
(N-ColE7), to decipher the role of the conserved asparagine and
histidine residues in the HNH motif. We found, using fluorescence
resonance energy transfer (FRET) assays, that the DNA hydrolysis
activity of H545 N-ColE7 mutants was completely abolished while
activities of N560 and H573 mutants varied from 6.9% to 83.2% of the
wild-type activity. The crystal structures of three N-ColE7 mutants in
complex with the inhibitor Im7, N560A-Im7, N560D-Im7 and H573A-Im7,
were determined at a resolution of 1.9 A to 2.2 A. H573 is responsible
for metal ion binding in the wild-type protein, as the zinc ion is
still partially associated in the structure of H573A, suggesting that
H573 plays a supportive role in metal binding. Both N560A and N560D
contain a disordered loop in the HNH motif due to the disruption of the
hydrogen bond network surrounding the side-chain of residue 560, and as
a result, the imidazole ring of the general base residue H545 is tilted
slightly and the scissile phosphate is shifted, leading to the large
reductions in hydrolysis activities. These results suggest that the
highly conserved asparagine in the HNH motif, in general, plays a
structural role in constraining the loop in the metal finger structure
and keeping the general base histidine and scissile phosphate in the
correct position for DNA hydrolysis.
Structural Basis for
Sequence-Dependent DNA Cleavage by Nonspecific Endonucleases.
(2007) Nucleic Acids Res.35:
584
PubMed Abstract:
Nonspecific endonucleases hydrolyze DNA without sequence specificity
but with sequence preference, however the structural basis for cleavage
preference remains elusive. We show here that the nonspecific
endonuclease ColE7 cleaves DNA with a preference for making nicks after
(at 3'O-side) thymine bases but the periplasmic nuclease Vvn cleaves
DNA more evenly with little sequence preference. The crystal structure
of the 'preferred complex' of the nuclease domain of ColE7 bound to an
18 bp DNA with a thymine before the scissile phosphate had a more
distorted DNA phosphate backbone than the backbones in the
non-preferred complexes, so that the scissile phosphate was
compositionally closer to the endonuclease active site resulting in
more efficient DNA cleavage. On the other hand, in the crystal
structure of Vvn in complex with a 16 bp DNA, the DNA phosphate
backbone was similar and not distorted in comparison with that of a
previously reported complex of Vvn with a different DNA sequence. Taken
together these results suggest a general structural basis for the
sequence-dependent DNA cleavage catalyzed by nonspecific endonucleases,
indicating that nonspecific nucleases could induce DNA to deform to
distinctive levels depending on the local sequence leading to different
cleavage rates along the DNA chain.
Crystal structural analysis and
metal-dependent stability and activity studies of the ColE7
endonuclease domain in complex with DNA/Zn2+ or inhibitor/Ni2+
(2006) Protein Sci.15:
269-280
PubMed Abstract:
The nuclease domain of ColE7 (N-ColE7) contains an H-N-H motif that
folds in a beta beta alpha-metal topology. Here we report the crystal
structures of a Zn2+-bound N-ColE7 (H545E mutant) in complex with a
12-bp duplex DNA and a Ni2+-bound N-ColE7 in complex with the inhibitor
Im7 at a resolution of 2.5 A and 2.0 A, respectively. Metal-dependent
cleavage assays showed that N-ColE7 cleaves double-stranded DNA with a
single metal ion cofactor, Ni2+, Mg2+, Mn2+, and Zn2+. ColE7 purified
from Escherichia coli contains an endogenous zinc ion that was not
replaced by Mg2+ at concentrations of <25 mM, indicating that
zinc is the physiologically relevant metal ion in N-ColE7 in host E.
coli. In the crystal structure of N-ColE7/DNA complex, the zinc ion is
directly coordinated to three histidines and the DNA scissile phosphate
in a tetrahedral geometry. In contrast, Ni2+ is bound in N-ColE7 in two
different modes, to four ligands (three histidines and one phosphate
ion), or to five ligands with an additional water molecule. These data
suggest that the divalent metal ion in the His-metal finger motif can
be coordinated to six ligands, such as Mg2+ in I-PpoI, Serratia
nuclease and Vvn, five ligands or four ligands, such as Ni2+ or Zn2+ in
ColE7. Universally, the metal ion in the His-metal finger motif is
bound to the DNA scissile phosphate and serves three roles during
hydrolysis: polarization of the P-O bond for nucleophilic attack,
stabilization of the phosphoanion transition state and stabilization of
the cleaved product.
Crystal structural analysis and
metal-dependent stability and activity studies of the ColE7
endonuclease domain in complex with DNA/Zn2+ or inhibitor/Ni2+
(2006) Protein Sci.15:
269-280
PubMed Abstract:
The nuclease domain of ColE7 (N-ColE7) contains an H-N-H motif that
folds in a beta beta alpha-metal topology. Here we report the crystal
structures of a Zn2+-bound N-ColE7 (H545E mutant) in complex with a
12-bp duplex DNA and a Ni2+-bound N-ColE7 in complex with the inhibitor
Im7 at a resolution of 2.5 A and 2.0 A, respectively. Metal-dependent
cleavage assays showed that N-ColE7 cleaves double-stranded DNA with a
single metal ion cofactor, Ni2+, Mg2+, Mn2+, and Zn2+. ColE7 purified
from Escherichia coli contains an endogenous zinc ion that was not
replaced by Mg2+ at concentrations of <25 mM, indicating that
zinc is the physiologically relevant metal ion in N-ColE7 in host E.
coli. In the crystal structure of N-ColE7/DNA complex, the zinc ion is
directly coordinated to three histidines and the DNA scissile phosphate
in a tetrahedral geometry. In contrast, Ni2+ is bound in N-ColE7 in two
different modes, to four ligands (three histidines and one phosphate
ion), or to five ligands with an additional water molecule. These data
suggest that the divalent metal ion in the His-metal finger motif can
be coordinated to six ligands, such as Mg2+ in I-PpoI, Serratia
nuclease and Vvn, five ligands or four ligands, such as Ni2+ or Zn2+ in
ColE7. Universally, the metal ion in the His-metal finger motif is
bound to the DNA scissile phosphate and serves three roles during
hydrolysis: polarization of the P-O bond for nucleophilic attack,
stabilization of the phosphoanion transition state and stabilization of
the cleaved product.
High-resolution crystal structure of
a truncated ColE7 translocation domain: implications for colicin
transport across membranes
(2006) J.Mol.Biol.356: 22-31
PubMed Abstract:
ColE7 is a nuclease-type colicin released from Escherichia coli to kill
sensitive bacterial cells by degrading the nucleic acid molecules in
their cytoplasm. ColE7 is classified as one of the group A colicins,
since the N-terminal translocation domain (T-domain) of the
nuclease-type colicins interact with specific membrane-bound or
periplasmic Tol proteins during protein import. Here, we show that if
the N-terminal tail of ColE7 is deleted, ColE7 (residues 63-576) loses
its bactericidal activity against E.coli. Moreover, TolB protein
interacts directly with the T-domain of ColE7 (residues 1-316), but not
with the N-terminal deleted T-domain (residues 60-316), as detected by
co-immunoprecipitation experiments, confirming that the N-terminal tail
is required for ColE7 interactions with TolB. The crystal structure of
the N-terminal tail deleted ColE7 T-domain was determined by the
multi-wavelength anomalous dispersion method at a resolution of 1.7
angstroms. The structure of the ColE7 T-domain superimposes well with
the T-domain of ColE3 and TR-domain of ColB, a group A Tol-dependent
colicin and a group B TonB-dependent colicin, respectively. The
structural resemblance of group A and B colicins implies that the two
groups of colicins may share a mechanistic connection during cellular
import.
DNA binding and degradation by the
HNH protein ColE7.
(2004) STRUCTURE12: 205-214
PubMed Abstract:
The bacterial toxin ColE7 bears an HNH motif which has been identified
in hundreds of prokaryotic and eukaryotic endonucleases, involved in
DNA homing, restriction, repair, or chromosome degradation. The crystal
structure of the nuclease domain of ColE7 in complex with a duplex DNA
has been determined at 2.5 A resolution. The HNH motif is bound at the
minor groove primarily to DNA phosphate groups at and beyond the 3'
side of the scissile phosphate, with little interaction with ribose
groups and bases. This result provides a structural basis for sugar-
and sequence-independent DNA recognition and the inhibition mechanism
by inhibitor Im7, which blocks the substrate binding site but not the
active site. Structural comparison shows that two families of
endonucleases bind and bend DNA in a similar way to that of the HNH
ColE7, indicating that endonucleases containing a "betabetaalpha-metal"
fold of active site possess a universal mode for protein-DNA
interactions.
Metal ions and phosphate binding in
the H-N-H motif: crystal structures of the nuclease domain of ColE7/Im7
in complex with a phosphate ion and different divalent metal ions
(2002) PROTEIN SCI.11:
2947-2957
PubMed Abstract:
H-N-H is a motif found in the nuclease domain of a subfamily of
bacteria toxins, including colicin E7, that are capable of cleaving DNA
nonspecifically. This H-N-H motif has also been identified in a
subfamily of homing endonucleases, which cleave DNA site specifically.
To better understand the role of metal ions in the H-N-H motif during
DNA hydrolysis, we crystallized the nuclease domain of colicin E7
(nuclease-ColE7) in complex with its inhibitor Im7 in two different
crystal forms, and we resolved the structures of EDTA-treated,
Zn(2+)-bound and Mn(2+)-bound complexes in the presence of phosphate
ions at resolutions of 2.6 A to 2.0 A. This study offers the first
determination of the structure of a metal-free and substrate-free
enzyme in the H-N-H family. The H-N-H motif contains two antiparallel
beta-strands linked to a C-terminal alpha-helix, with a divalent metal
ion located in the center. Here we show that the metal-binding sites in
the center of the H-N-H motif, for the EDTA-treated and Mg(2+)-soaked
complex crystals, were occupied by water molecules, indicating that an
alkaline earth metal ion does not reside in the same position as a
transition metal ion in the H-N-H motif. However, a Zn(2+) or Mn(2+)
ions were observed in the center of the H-N-H motif in cases of Zn(2+)
or Mn(2+)-soaked crystals, as confirmed in anomalous difference maps. A
phosphate ion was found to bridge between the divalent transition metal
ion and His545. Based on these structures and structural comparisons
with other nucleases, we suggest a functional role for the divalent
transition metal ion in the H-N-H motif in stabilizing the phosphoanion
in the transition state during hydrolysis.
The Crystal Structure of the
Nuclease Domain of Colicin E7 Suggests a Mechanism for Binding to
Double-stranded DNA by the H-N-H Endonucleases
(2002) J.mol.biol.324: 227-236
PubMed Abstract:
The bacterial toxin ColE7 contains an H-N-H endonuclease domain
(nuclease ColE7) that digests cellular DNA or RNA non-specifically in
target cells, leading to cell death. In the host cell, protein Im7
forms a complex with ColE7 to inhibit its nuclease activity. Here, we
present the crystal structure of the unbound nuclease ColE7 at a
resolution of 2.1A. Structural comparison between the unbound and bound
nuclease ColE7 in complex with Im7, suggests that Im7 is not an
allosteric inhibitor that induces backbone conformational changes in
nuclease ColE7, but rather one that inhibits by blocking the
substrate-binding site. There were two nuclease ColE7 molecules in the
P1 unit cell in crystals and they appeared as a dimer related to each
other by a non-crystallographic dyad symmetry. Gel-filtration and
cross-linking experiments confirmed that nuclease ColE7 indeed formed
dimers in solution and that the dimeric conformation was more favored
in the presence of double-stranded DNA. Structural comparison of
nuclease ColE7 with the His-Cys box homing endonuclease I-PpoI further
demonstrated that H-N-H motifs in dimeric nuclease ColE7 were oriented
in a manner very similar to that of the betabetaalpha-fold of the
active sites found in dimeric I-PpoI. A mechanism for the binding of
double-stranded DNA by dimeric H-N-H nuclease ColE7 is suggested.
The crystal structure of the
immunity protein of colicin E7 suggests a possible colicin-interacting
surface.
(1996) Proc.Natl.Acad.Sci.USA93:
6437-6442
PubMed Abstract:
The immunity protein of colicin E7 (ImmE7) can bind specifically to the
DNase-type colicin E7 and inhibit its bactericidal activity. Here we
report the 1.8-angstrom crystal structure of the ImmE7 protein. This is
the first x-ray structure determined in the superfamily of colicin
immunity proteins. The ImmE7 protein consists of four antiparallel
alpha-helices, folded in a topology similar to the architecture of a
four-helix bundle structure. A region rich in acidic residues is
identified. This negatively charged area has the greatest variability
within the family of DNase-type immunity proteins; thus, it seems
likely that this area is involved in specific binding to colicin. Based
on structural, genetic, and kinetic data, we suggest that all the
DNase-type immunity proteins, as well as colicins, share a
"homologous-structural framework" and that specific interaction between
a colicin and its cognate immunity protein relies upon how well these
two proteins' charged residues match on the interaction surface, thus
leading to specific immunity of the colicin.
Structural Basis for
Sequence-Dependent DNA Cleavage by Nonspecific Endonucleases.
(2007) Nucleic Acids Res.35:
584
PubMed Abstract:
Nonspecific endonucleases hydrolyze DNA without sequence specificity
but with sequence preference, however the structural basis for cleavage
preference remains elusive. We show here that the nonspecific
endonuclease ColE7 cleaves DNA with a preference for making nicks after
(at 3'O-side) thymine bases but the periplasmic nuclease Vvn cleaves
DNA more evenly with little sequence preference. The crystal structure
of the 'preferred complex' of the nuclease domain of ColE7 bound to an
18 bp DNA with a thymine before the scissile phosphate had a more
distorted DNA phosphate backbone than the backbones in the
non-preferred complexes, so that the scissile phosphate was
compositionally closer to the endonuclease active site resulting in
more efficient DNA cleavage. On the other hand, in the crystal
structure of Vvn in complex with a 16 bp DNA, the DNA phosphate
backbone was similar and not distorted in comparison with that of a
previously reported complex of Vvn with a different DNA sequence. Taken
together these results suggest a general structural basis for the
sequence-dependent DNA cleavage catalyzed by nonspecific endonucleases,
indicating that nonspecific nucleases could induce DNA to deform to
distinctive levels depending on the local sequence leading to different
cleavage rates along the DNA chain.
DNA binding and cleavage by the
periplasmic nuclease Vvn: a novel structure with a known active site.
(2003) Embo J.22: 4014-4025
PubMed Abstract:
The Vibrio vulnificus nuclease, Vvn, is a non-specific periplasmic
nuclease capable of digesting DNA and RNA. The crystal structure of Vvn
and that of Vvn mutant H80A in complex with DNA were resolved at 2.3 A
resolution. Vvn has a novel mixed alpha/beta topology containing four
disulfide bridges, suggesting that Vvn is not active under reducing
conditions in the cytoplasm. The overall structure of Vvn shows no
similarity to other endonucleases; however, a known
'betabetaalpha-metal' motif is identified in the central cleft region.
The crystal structure of the mutant Vvn-DNA complex demonstrates that
Vvn binds mainly at the minor groove of DNA, resulting in duplex
bending towards the major groove by approximately 20 degrees. Only the
DNA phosphate backbones make hydrogen bonds with Vvn, suggesting a
structural basis for its sequence-independent recognition of DNA and
RNA. Based on the enzyme-substrate and enzyme-product structures
observed in the mutant Vvn-DNA crystals, a catalytic mechanism is
proposed. This structural study suggests that Vvn hydrolyzes DNA by a
general single-metal ion mechanism, and indicates how non-specific
DNA-binding proteins may recognize DNA.
DNA binding and cleavage by the
periplasmic nuclease Vvn: a novel structure with a known active site.
(2003) Embo J.22: 4014-4025
PubMed Abstract:
The Vibrio vulnificus nuclease, Vvn, is a non-specific periplasmic
nuclease capable of digesting DNA and RNA. The crystal structure of Vvn
and that of Vvn mutant H80A in complex with DNA were resolved at 2.3 A
resolution. Vvn has a novel mixed alpha/beta topology containing four
disulfide bridges, suggesting that Vvn is not active under reducing
conditions in the cytoplasm. The overall structure of Vvn shows no
similarity to other endonucleases; however, a known
'betabetaalpha-metal' motif is identified in the central cleft region.
The crystal structure of the mutant Vvn-DNA complex demonstrates that
Vvn binds mainly at the minor groove of DNA, resulting in duplex
bending towards the major groove by approximately 20 degrees. Only the
DNA phosphate backbones make hydrogen bonds with Vvn, suggesting a
structural basis for its sequence-independent recognition of DNA and
RNA. Based on the enzyme-substrate and enzyme-product structures
observed in the mutant Vvn-DNA crystals, a catalytic mechanism is
proposed. This structural study suggests that Vvn hydrolyzes DNA by a
general single-metal ion mechanism, and indicates how non-specific
DNA-binding proteins may recognize DNA.
Crystal Structure of a Natural
Circularly Permuted Jellyroll Protein: 1,3-1,4-beta-D-Glucanase from
Fibrobacter succinogenes.
(2003) J.Mol.Biol.330: 607-620
PubMed Abstract:
The 1,3-1,4-beta-D-glucanase from Fibrobacter succinogenes
(Fsbeta-glucanase) is classified as one of the family 16 glycosyl
hydrolases. It hydrolyzes the glycosidic bond in the mixed-linked
glucans containing beta-1,3- and beta-1,4-glycosidic linkages. We
constructed a truncated form of recombinant Fsbeta-glucanase containing
the catalytic domain from amino acid residues 1-258, which exhibited a
higher thermal stability and enzymatic activity than the full-length
enzyme. The crystal structure of the truncated Fsbeta-glucanase was
solved at a resolution of 1.7A by the multiple wavelength anomalous
dispersion (MAD) method using the anomalous signals from the
seleno-methionine-labeled protein. The overall topology of the
truncated Fsbeta-glucanase consists mainly of two eight-stranded
anti-parallel beta-sheets arranged in a jellyroll beta-sandwich,
similar to the fold of many glycosyl hydrolases and
carbohydrate-binding modules. Sequence comparison with other bacterial
glucanases showed that Fsbeta-glucanase is the only naturally occurring
circularly permuted beta-glucanase with reversed sequences. Structural
comparison shows that the engineered circular-permuted Bacillus enzymes
are more similar to their parent enzymes with which they share
approximately 70% sequence identity, than to the naturally occurring
Fsbeta-glucanase of similar topology with 30% identity. This result
suggests that protein structure relies more on sequence identity than
topology. The high-resolution structure of Fsbeta-glucanase provides a
structural rationale for the different activities obtained from a
series of mutant glucanases and a basis for the development of
engineered enzymes with increased activity and structural stability.
Structural analysis of the
transcriptional activation on Fis: crystal structures of six Fis
mutants with different activation properties.
(2000) J.Mol.Biol.302: 1139-1151
PubMed Abstract:
The Fis protein regulates gene expression in Escherichia coli by
activating or repressing transcription of a variety of genes. Fis can
activate transcription when bound to DNA upstream of the
RNA-polymerase-binding site, such as in the rrnB P1 promoter, or when
bound to a site overlapping the -35 RNA polymerase binding site, such
as in the proP P2 promoter. It has been suggested that transcriptional
activation in both promoters results from interactions between specific
amino acids within a turn connecting the B and C helices (the BC turn)
in Fis and the C-terminal domain of the alpha-subunit of RNA polymerase
(alphaCTD of RNAP). Here, crystal structures of six Fis BC turn mutants
with different transcriptional activation properties, Q68A, R71Y, R71L,
G72A, G72D and Q74A, were determined at 1.9 to 2.8 A resolution. Two of
these mutants, R71Y and R71L, crystallized in unit cells which are
different from that of wild-type Fis, and the structure of R71L offers
the most complete Fis model to date in that the extended structure of
the N-terminal region is revealed. The BC turn in all of these mutant
structures remains in a nearly identical gamma gamma beta-turn
conformation as present in wild-type Fis. Analyses of the molecular
surfaces of the transactivation region of the mutants suggest that
several residues in or near the BC turn, including Gln68, Arg71, Gly72
and Gln74, form a ridge that could contact the alphaCTD of RNAP on one
side. The structures and biochemical properties of the mutants suggest
that Arg71 is the most critical residue for contacting RNAP within this
ridge and that the glycine at position 72 helps to stabilize the
structure.
Structural analysis of the
transcriptional activation on Fis: crystal structures of six Fis
mutants with different activation properties.
(2000) J.Mol.Biol.302: 1139-1151
PubMed Abstract:
The Fis protein regulates gene expression in Escherichia coli by
activating or repressing transcription of a variety of genes. Fis can
activate transcription when bound to DNA upstream of the
RNA-polymerase-binding site, such as in the rrnB P1 promoter, or when
bound to a site overlapping the -35 RNA polymerase binding site, such
as in the proP P2 promoter. It has been suggested that transcriptional
activation in both promoters results from interactions between specific
amino acids within a turn connecting the B and C helices (the BC turn)
in Fis and the C-terminal domain of the alpha-subunit of RNA polymerase
(alphaCTD of RNAP). Here, crystal structures of six Fis BC turn mutants
with different transcriptional activation properties, Q68A, R71Y, R71L,
G72A, G72D and Q74A, were determined at 1.9 to 2.8 A resolution. Two of
these mutants, R71Y and R71L, crystallized in unit cells which are
different from that of wild-type Fis, and the structure of R71L offers
the most complete Fis model to date in that the extended structure of
the N-terminal region is revealed. The BC turn in all of these mutant
structures remains in a nearly identical gamma gamma beta-turn
conformation as present in wild-type Fis. Analyses of the molecular
surfaces of the transactivation region of the mutants suggest that
several residues in or near the BC turn, including Gln68, Arg71, Gly72
and Gln74, form a ridge that could contact the alphaCTD of RNAP on one
side. The structures and biochemical properties of the mutants suggest
that Arg71 is the most critical residue for contacting RNAP within this
ridge and that the glycine at position 72 helps to stabilize the
structure.
Structural analysis of the
transcriptional activation on Fis: crystal structures of six Fis
mutants with different activation properties.
(2000) J.Mol.Biol.302: 1139-1151
PubMed Abstract:
The Fis protein regulates gene expression in Escherichia coli by
activating or repressing transcription of a variety of genes. Fis can
activate transcription when bound to DNA upstream of the
RNA-polymerase-binding site, such as in the rrnB P1 promoter, or when
bound to a site overlapping the -35 RNA polymerase binding site, such
as in the proP P2 promoter. It has been suggested that transcriptional
activation in both promoters results from interactions between specific
amino acids within a turn connecting the B and C helices (the BC turn)
in Fis and the C-terminal domain of the alpha-subunit of RNA polymerase
(alphaCTD of RNAP). Here, crystal structures of six Fis BC turn mutants
with different transcriptional activation properties, Q68A, R71Y, R71L,
G72A, G72D and Q74A, were determined at 1.9 to 2.8 A resolution. Two of
these mutants, R71Y and R71L, crystallized in unit cells which are
different from that of wild-type Fis, and the structure of R71L offers
the most complete Fis model to date in that the extended structure of
the N-terminal region is revealed. The BC turn in all of these mutant
structures remains in a nearly identical gamma gamma beta-turn
conformation as present in wild-type Fis. Analyses of the molecular
surfaces of the transactivation region of the mutants suggest that
several residues in or near the BC turn, including Gln68, Arg71, Gly72
and Gln74, form a ridge that could contact the alphaCTD of RNAP on one
side. The structures and biochemical properties of the mutants suggest
that Arg71 is the most critical residue for contacting RNAP within this
ridge and that the glycine at position 72 helps to stabilize the
structure.
Structural analysis of the
transcriptional activation on Fis: crystal structures of six Fis
mutants with different activation properties.
(2000) J.Mol.Biol.302: 1139-1151
PubMed Abstract:
The Fis protein regulates gene expression in Escherichia coli by
activating or repressing transcription of a variety of genes. Fis can
activate transcription when bound to DNA upstream of the
RNA-polymerase-binding site, such as in the rrnB P1 promoter, or when
bound to a site overlapping the -35 RNA polymerase binding site, such
as in the proP P2 promoter. It has been suggested that transcriptional
activation in both promoters results from interactions between specific
amino acids within a turn connecting the B and C helices (the BC turn)
in Fis and the C-terminal domain of the alpha-subunit of RNA polymerase
(alphaCTD of RNAP). Here, crystal structures of six Fis BC turn mutants
with different transcriptional activation properties, Q68A, R71Y, R71L,
G72A, G72D and Q74A, were determined at 1.9 to 2.8 A resolution. Two of
these mutants, R71Y and R71L, crystallized in unit cells which are
different from that of wild-type Fis, and the structure of R71L offers
the most complete Fis model to date in that the extended structure of
the N-terminal region is revealed. The BC turn in all of these mutant
structures remains in a nearly identical gamma gamma beta-turn
conformation as present in wild-type Fis. Analyses of the molecular
surfaces of the transactivation region of the mutants suggest that
several residues in or near the BC turn, including Gln68, Arg71, Gly72
and Gln74, form a ridge that could contact the alphaCTD of RNAP on one
side. The structures and biochemical properties of the mutants suggest
that Arg71 is the most critical residue for contacting RNAP within this
ridge and that the glycine at position 72 helps to stabilize the
structure.
Structural analysis of the
transcriptional activation on Fis: crystal structures of six Fis
mutants with different activation properties.
(2000) J.Mol.Biol.302: 1139-1151
PubMed Abstract:
The Fis protein regulates gene expression in Escherichia coli by
activating or repressing transcription of a variety of genes. Fis can
activate transcription when bound to DNA upstream of the
RNA-polymerase-binding site, such as in the rrnB P1 promoter, or when
bound to a site overlapping the -35 RNA polymerase binding site, such
as in the proP P2 promoter. It has been suggested that transcriptional
activation in both promoters results from interactions between specific
amino acids within a turn connecting the B and C helices (the BC turn)
in Fis and the C-terminal domain of the alpha-subunit of RNA polymerase
(alphaCTD of RNAP). Here, crystal structures of six Fis BC turn mutants
with different transcriptional activation properties, Q68A, R71Y, R71L,
G72A, G72D and Q74A, were determined at 1.9 to 2.8 A resolution. Two of
these mutants, R71Y and R71L, crystallized in unit cells which are
different from that of wild-type Fis, and the structure of R71L offers
the most complete Fis model to date in that the extended structure of
the N-terminal region is revealed. The BC turn in all of these mutant
structures remains in a nearly identical gamma gamma beta-turn
conformation as present in wild-type Fis. Analyses of the molecular
surfaces of the transactivation region of the mutants suggest that
several residues in or near the BC turn, including Gln68, Arg71, Gly72
and Gln74, form a ridge that could contact the alphaCTD of RNAP on one
side. The structures and biochemical properties of the mutants suggest
that Arg71 is the most critical residue for contacting RNAP within this
ridge and that the glycine at position 72 helps to stabilize the
structure.
Structural analysis of the
transcriptional activation on Fis: crystal structures of six Fis
mutants with different activation properties.
(2000) J.Mol.Biol.302: 1139-1151
PubMed Abstract:
The Fis protein regulates gene expression in Escherichia coli by
activating or repressing transcription of a variety of genes. Fis can
activate transcription when bound to DNA upstream of the
RNA-polymerase-binding site, such as in the rrnB P1 promoter, or when
bound to a site overlapping the -35 RNA polymerase binding site, such
as in the proP P2 promoter. It has been suggested that transcriptional
activation in both promoters results from interactions between specific
amino acids within a turn connecting the B and C helices (the BC turn)
in Fis and the C-terminal domain of the alpha-subunit of RNA polymerase
(alphaCTD of RNAP). Here, crystal structures of six Fis BC turn mutants
with different transcriptional activation properties, Q68A, R71Y, R71L,
G72A, G72D and Q74A, were determined at 1.9 to 2.8 A resolution. Two of
these mutants, R71Y and R71L, crystallized in unit cells which are
different from that of wild-type Fis, and the structure of R71L offers
the most complete Fis model to date in that the extended structure of
the N-terminal region is revealed. The BC turn in all of these mutant
structures remains in a nearly identical gamma gamma beta-turn
conformation as present in wild-type Fis. Analyses of the molecular
surfaces of the transactivation region of the mutants suggest that
several residues in or near the BC turn, including Gln68, Arg71, Gly72
and Gln74, form a ridge that could contact the alphaCTD of RNAP on one
side. The structures and biochemical properties of the mutants suggest
that Arg71 is the most critical residue for contacting RNAP within this
ridge and that the glycine at position 72 helps to stabilize the
structure.
Structural analysis of the
transcriptional activation on Fis: crystal structures of six Fis
mutants with different activation properties.
(2000) J.Mol.Biol.302: 1139-1151
PubMed Abstract:
The Fis protein regulates gene expression in Escherichia coli by
activating or repressing transcription of a variety of genes. Fis can
activate transcription when bound to DNA upstream of the
RNA-polymerase-binding site, such as in the rrnB P1 promoter, or when
bound to a site overlapping the -35 RNA polymerase binding site, such
as in the proP P2 promoter. It has been suggested that transcriptional
activation in both promoters results from interactions between specific
amino acids within a turn connecting the B and C helices (the BC turn)
in Fis and the C-terminal domain of the alpha-subunit of RNA polymerase
(alphaCTD of RNAP). Here, crystal structures of six Fis BC turn mutants
with different transcriptional activation properties, Q68A, R71Y, R71L,
G72A, G72D and Q74A, were determined at 1.9 to 2.8 A resolution. Two of
these mutants, R71Y and R71L, crystallized in unit cells which are
different from that of wild-type Fis, and the structure of R71L offers
the most complete Fis model to date in that the extended structure of
the N-terminal region is revealed. The BC turn in all of these mutant
structures remains in a nearly identical gamma gamma beta-turn
conformation as present in wild-type Fis. Analyses of the molecular
surfaces of the transactivation region of the mutants suggest that
several residues in or near the BC turn, including Gln68, Arg71, Gly72
and Gln74, form a ridge that could contact the alphaCTD of RNAP on one
side. The structures and biochemical properties of the mutants suggest
that Arg71 is the most critical residue for contacting RNAP within this
ridge and that the glycine at position 72 helps to stabilize the
structure.
The crystal structure of the DNase
domain of colicin E7 in complex with its inhibitor Im7 protein.
(1999) Structure Fold.Des.7:
91-102
PubMed Abstract:
Colicin E7 (ColE7) is one of the bacterial toxins classified as a
DNase-type E-group colicin. The cytotoxic activity of a colicin in a
colicin-producing cell can be counteracted by binding of the colicin to
a highly specific immunity protein. This biological event is a good
model system for the investigation of protein recognition.
Crystallization and preliminary
crystallographic analysis of the Escherichia coli tyrosine
aminotransferase.
(1999) Acta Crystallogr.,Sect.D55:
1474-1477
PubMed Abstract:
Tyrosine aminotransferase catalyzes transamination for both
dicarboxylic and aromatic amino-acid substrates. The substrate-free
Escherichia coli tyrosine aminotransferase (eTAT) bound with the
cofactor pyridoxal 5'-phosphate (PLP) was crystallized in the trigonal
space group P3(2). A low-resolution crystal structure of eTAT was
determined by molecular-replacement methods. The overall folding of
eTAT resembles that of the aspartate aminotransferases, with the two
identical subunits forming a dimer in which each monomer binds a PLP
molecule via a covalent bond linked to the epsilon-NH(2) group of
Lys258. Comparison of the structure of eTAT with those of the open,
half-open or closed form of chicken or E. coli aspartate
aminotransferases shows the eTAT structure to be in the open
conformation.
A novel role of ImmE7 in the
autoregulatory expression of the ColE7 operon and identification of
possible RNase active sites in the crystal structure of dimeric ImmE7.
(1997) EMBO J.16: 1444-1454
PubMed Abstract:
Site-specific cleavage of mRNA has been identified in vivo for the
polycistronic colicin E7 operon (ColE7), which occurs between G and A
nucleotides located at the Asp52 codon (GAT) of the immunity gene
(ceiE7). In vitro, this specific cleavage occurs only in the presence
of the ceiE7 gene product (ImmE7). The crystal structure of dimeric
ImmE7 has been determined at 1.8 A resolution by X-ray crystallographic
analysis. We found that several residues located at the interface of
dimeric ImmE7 bear surprising resemblance to the active sites of some
RNases. These results suggest that dimeric ImmE7 may possess a novel
RNase activity that cleaves its own mRNA at a specific site and thus
autoregulates translational expression of the downstream celE7 gene as
well as degradation of the upstream ceaE7 mRNA.
The transactivation region of the
fis protein that controls site-specific DNA inversion contains extended
mobile beta-hairpin arms.
(1997) EMBO J.16: 6860-6873
PubMed Abstract:
The Fis protein regulates site-specific DNA inversion catalyzed by a
family of DNA invertases when bound to a cis-acting recombinational
enhancer. As is often found for transactivation domains, previous
crystal structures have failed to resolve the conformation of the
N-terminal inversion activation region within the Fis dimer. A new
crystal form of a mutant Fis protein now reveals that the activation
region contains two beta-hairpin arms that protrude over 20 A from the
protein core. Saturation mutagenesis identified the regulatory and
structurally important amino acids. The most critical activating
residues are located near the tips of the beta-arms. Disulfide
cross-linking between the beta-arms demonstrated that they are highly
flexible in solution and that efficient inversion activation can occur
when the beta-arms are covalently linked together. The emerging picture
for this regulatory motif is that contacts with the recombinase at the
tip of the mobile beta-arms activate the DNA invertase in the context
of an invertasome complex.
UNKNOWN PEPTIDE, POSSIBLY PART OF THE
UNOBSERVED RESIDUES IN ENTITY 1
Polymer:
2
Type:
protein
Length:
4
Chains:
C, D
Citation:
The structure of Fis mutant Pro61Ala
illustrates that the kink within the long alpha-helix is not due to the
presence of the proline residue.
(1994) J.Biol.Chem.269:
28947-28954
PubMed Abstract:
The influence of proline on bending of the alpha-helix was investigated
by replacement of the proline residue located in the middle of the long
alpha-helix of the Fis protein with alanine, serine, or leucine. Each
of the three substitutions folded into a stable protein with the same
or higher melting points than the wild-type, but only Pro61Ala was
functionally active in stimulating Hin-mediated DNA inversion. Pro61Ala
formed crystals that were isomorphous with the wild-type protein
allowing the structure to be determined at 1.9-A resolution by x-ray
diffraction methods. The structure of the Pro61Ala mutant is almost
identical to the wild-type protein, consistent with its near wild-type
activity. One of the alpha-helices, the B-helix, is kinked in the
wild-type Fis protein by 20 degrees which was previously assumed to be
caused solely by the presence of proline 61 in the center of the helix.
However, the B-helix is still kinked by 16 degrees when proline 61 is
replaced by alanine. Local peptide backbone movement around residue 57
adjusts the geometry of the helix to accommodate the new main chain
hydrogen bond between the -CO group in Glu57 and the -NH group in
Ala61. Thus, the kink of the alpha-helix in Pro61Ala does not require
the presence of proline.
The molecular structure of wild-type
and a mutant Fis protein: relationship between mutational changes and
recombinational enhancer function or DNA binding.
(1991) Proc.Natl.Acad.Sci.USA88:
9558-9562
PubMed Abstract:
The 98-amino acid Fis protein from Escherichia coli functions in a
variety of reactions, including promotion of Hin-mediated site-specific
DNA inversion when bound to an enhancer sequence. It is unique among
site-specific DNA-binding proteins in that it binds to a large number
of different DNA sequences, for which a consensus sequence is difficult
to establish. X-ray crystal structure analyses have been carried out at
2.3 A resolution for wild-type Fis and for an Arg-89----Cys mutant that
does not stimulate DNA inversion. Each monomer of the Fis dimer has
four alpha-helices, A-D; the first 19 residues are disordered in the
crystal. The end of each C helix is hydrogen bonded to the beginning of
helix B' from the opposite subunit in what effectively is one long
continuous, although bent, helix. The four helices, C, B', C', and B,
together define a platform through the center of the Fis molecule:
helices A and A' are believed to be involved with Hin recombinase on
one side, and helices D and D' interact with DNA lying on the other
side of the platform. Helices C and D of each subunit comprise a
helix-turn-helix (HTH) DNA-binding element. The spacing of these two
HTH elements in the dimer, 25 A, is too short to allow insertion into
adjacent major grooves of a straight B-DNA helix. However, bending the
DNA at discrete points, to an overall radius of curvature of 62 A,
allows efficient docking of a B-DNA helix with the Fis molecule. The
proposed complex explains the experimentally observed patterns of
methylation protection and DNase I cleavage hypersensitivity. The x-ray
structure accounts for the effects of mutations in the Fis sequence.
Those that affect DNA inversion but not DNA binding are located within
the N-terminal disordered region and helix A. This inversion activation
domain is physically separated in the Fis molecule from the HTH
elements and may specify a region of contact with the Hin recombinase.
In contrast, mutations that affect HTH helices C and D, or interactions
of these with helix B, have the additional effect of decreasing or
eliminating binding to DNA.
The molecular structure of wild-type
and a mutant Fis protein: relationship between mutational changes and
recombinational enhancer function or DNA binding.
(1991) Proc.Natl.Acad.Sci.USA88:
9558-9562
PubMed Abstract:
The 98-amino acid Fis protein from Escherichia coli functions in a
variety of reactions, including promotion of Hin-mediated site-specific
DNA inversion when bound to an enhancer sequence. It is unique among
site-specific DNA-binding proteins in that it binds to a large number
of different DNA sequences, for which a consensus sequence is difficult
to establish. X-ray crystal structure analyses have been carried out at
2.3 A resolution for wild-type Fis and for an Arg-89----Cys mutant that
does not stimulate DNA inversion. Each monomer of the Fis dimer has
four alpha-helices, A-D; the first 19 residues are disordered in the
crystal. The end of each C helix is hydrogen bonded to the beginning of
helix B' from the opposite subunit in what effectively is one long
continuous, although bent, helix. The four helices, C, B', C', and B,
together define a platform through the center of the Fis molecule:
helices A and A' are believed to be involved with Hin recombinase on
one side, and helices D and D' interact with DNA lying on the other
side of the platform. Helices C and D of each subunit comprise a
helix-turn-helix (HTH) DNA-binding element. The spacing of these two
HTH elements in the dimer, 25 A, is too short to allow insertion into
adjacent major grooves of a straight B-DNA helix. However, bending the
DNA at discrete points, to an overall radius of curvature of 62 A,
allows efficient docking of a B-DNA helix with the Fis molecule. The
proposed complex explains the experimentally observed patterns of
methylation protection and DNase I cleavage hypersensitivity. The x-ray
structure accounts for the effects of mutations in the Fis sequence.
Those that affect DNA inversion but not DNA binding are located within
the N-terminal disordered region and helix A. This inversion activation
domain is physically separated in the Fis molecule from the HTH
elements and may specify a region of contact with the Hin recombinase.
In contrast, mutations that affect HTH helices C and D, or interactions
of these with helix B, have the additional effect of decreasing or
eliminating binding to DNA.